Zheng Wu1,
Wenzheng Li1,
Yuhua Sun2,
Kun Fu1,
Shujuan Cheng1 ![]()
For correspondence:- Shujuan Cheng Email: sjcheng6@163.com
Received: 25 October 2016 Accepted: 16 February 2017 Published: 31 March 2017
Citation: Wu Z, Li W, Sun Y, Fu K, Cheng S. Eleutheroside E inhibits doxorubicin-induced inflammation and apoptosis in rat cardiomyocytes by modulating activation of NF-κB pathway. Trop J Pharm Res 2017; 16(3):515-523 doi: 10.4314/tjpr.v16i3.4
© 2017 The authors.
This is an Open Access article that uses a funding model which does not charge readers or their institutions for access and distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0) and the Budapest Open Access Initiative (http://www.budapestopenaccessinitiative.org/read), which permit unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited..
Purpose: To identify the effects of eleutheroside E (EE) on apoptosis and inflammation induced by doxorubicin (DOX) in H9c2 cells and to investigate the underlying mechanisms.
Methods: The effect of EE on H9c2 cell viability was determined using Cell Counting Kit-8 (CCK8). EE effect on DOX-induced apoptosis and inflammation in H9c2 cells was studied by comparison between cells treated with DOX alone and DOX+EE; the relationship between EE effects and NF-κB signaling pathway was evaluated by the addition of NF-κB inhibitor PDTC. Cell apoptosis was examined by flow cytometry while IL-1β, IL-6, and TNF-α levels were determined by ELISA. The phosphorylation level of NF-κB p65 was measured by Western blot.
Results: Compared with control group, cell viability was notably elevated after treatment with 50-100 μM EE for 48 or 72 h. DOX induced higher rates of cell apoptosis in H9c2 cells (29.5 ± 3.56 %) compared with control group (6.39 ± 0.67 %); however, with EE pretreatment (50 and 80 μM), apoptosis rate decreased to 16.8 ± 2.16 and 13.54 ± 2.08 %, respectively, which are significantly lower than that of DOX group; furthermore, the levels of IL-1β, IL-6, and TNF-α also reduced. In addition, DOX-induced phosphorylation of NF-κB p65 was suppressed by EE pretreatment (10, 50 and 80 μM) to 11.51 ± 1.25, 40.2 ± 5.17 and 52.97 ± 6.74 %, respectively
Conclusion: The results suggest that EE treatment reduced DOX-induced apoptosis and inflammation by interacting with NF-κB signaling pathway. This finding sheds some light on probable new strategies on the application of DOX for cancer treatment
Introduction
Doxorubicin (DOX), one of the most commonly used anti-cancer drugs, possesses therapeutic effects for a variety of cancers, including leukemia, lymphoma, soft tissue sarcomas and solid tumors [1-4]. However, its clinical use is limited by a dose-related acute and chronic cardiotoxicity [5,6]. While the mechanism of DOX-induced cardiotoxicity still remains unclear, recent evidence have demonstrated that oxidative stress and cardiac inflammation were involved in the process [7-10]. Therefore 3-hydroxy-3-methylglutaryl coenzyme A (reductase inhibitor, which has anti-inflammatory and anti-oxidative effects) has been used to lessen the cardiotoxicity of DOX in mice [11]. In addition, both suppressed cardiac cytokine activation and lipid peroxidation have been shown to play critical roles in improving left ventricular (LV) function in a mouse model of DOX-induced cardiotoxicity [8,11].
Eleutheroside E (EE), an active component of Eleutherococcus senticosus, is known to exert an anti-inflammatory effect by inhibiting inflammatory cytokine expression [12,13]. However, the effects of EE on DOX-induced injury to cardiomyocytes and its underlying mechanism remain unclear.
In this study, we identified the effects of EE on H9c2 cell proliferation DOX-induced apoptosis and associated inflammation. Its effect on phosphorylation level of NF-κB p65 was also investigated to understand the role of NF-κB signaling pathway in the protective effect of EE.
Methods
Eleutheroside E
EE (CAS: 39432-56-9) white crystalline powder with a purity of ≥ 98 % was purchased from Chroma Biotechnology Co. Ltd. (Chengdu, China). It was dissolved in appropriate amount of dimethylsulfoxide (DMSO) and diluted to target concentrations before utilization, with the final DMSO concentrations below 0.5 %.
Cell culture, cell grouping and viability assay
H9c2 cardiac cells were cultured in 96-well plates containing DMEM-F12 medium supplemented with 10 % FBS and maintained at 37 °C/5 % CO2. To examine the effects of EE on DOX (Sigma-Aldrich, St. Louis, MO, USA) induced injury, H9c2 cells were divided into five groups: control, DOX(1 μM), EE (10 μM), EE (50 μM) and EE (80 μM) groups. In all EE groups, H9c2 cardiac cells were pretreated with various concentrations of EE (10, 50 and 80 μM) for 2 h prior to DOX (1 μM) treatment.
In order to further study whether the protective effects of EE were associated with the inhibition of NF-κB signaling, H9c2 cells were divided into six groups named as control, PDTC, DOX, EE (50 μM), EE (50 μM) + DOX and PDTC + DOX groups. PDTC group, DOX group, and EE group was treated with 10 μM PDTC, 1 μM DOX, and 50 μM EE, respectively. Cells in DOX + EE and PDTC +DOX groups were pretreated with EE (50 μM) or PDTC (10 μM) for 2 h prior to DOX treatment.
For viability assay, Cell Counting Kit-8 (CCK8) (Sigma-Aldrich, St. Louis, MO, USA) solution (10 μL) was added to each well at 1/10 dilution, followed by incubation of 2 hours. Absorbance was measured at 450 nm with a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA).
Determination of cell apoptosis
Cell apoptosis was detected with a flow cytometer (FCM) using an Annexin V-FITC apoptosis detection kit (Zhongze, Shanghai, China) following the manufacturer’s instructions. After DOX treatment, H9c2 cells were harvested, washed with cold PBS, and re-suspended in 500 μl binding buffer. Then 5μL of Annexin V stock solution was added and the cells were incubated for 10 min at 4°C. Propidium iodide (PI,5 μL/well) was added to the cells, which were immediately analyzed using a FACS-Calibur flow cytometer (Becton Dickinson, San Jose, CA, USA).
Analysis of nuclear morphology
H9c2 cells were cultured in a 6-well plate with a cell density of 2 × 105 per well for 24 h and pretreated with different doses of EE for 2 h prior to DOX treatment. Cells were harvested, rinsed twice with PBS, fixed with 1 mL of 4 % paraformaldehyde for 10 min, and then rinsed twice with PBS (5 min/time).
After being stained with 0.5 ml of Hoechst 33258 (Sigma, St. Louis, MO, USA) nuclear dye (10 μM) for 5 min, the cells were washed twice with PBS, then observed and photographed under an inverted fluorescence microscope (DMI3000B, Leica, Wetzlar, Germany) with the excitation and emission wavelengths of 340 and 460 nm, respectively. Apoptotic cells were identified on the basis of nuclear morphology changes, such as chromatin condensation and fragmentation.
ELISA
H92c cells were cultured in 96-well plates. After the indicated treatments, the levels of IL-1β, IL-6, and TNF-α in the culture media were measured using ELISA kits (Takara, Kusatsu, Japan). Each experiment was repeated at least 5 times.
Extraction of cytoplasmic and nuclear proteins
After the indicated treatment, H9c2 cells were harvested. Proteins in cytoplasm and nuclei from H9c2 cells were isolated by the NE-PER Nuclear and Cytoplasmic Extraction Kit according to the manufacturer’s protocol (Thermo Fisher Scientific, Waltham, MA, USA). The cytoplasmic and nuclear protein extracts were prepared for Western blot analysis.
Western blot
After the indicated treatments, H9c2 cells were harvested, lysed, and the homogenate was centrifuged at 12,000 rpm for 10 min at 4 oC. The protein concentration in the supernatant was determined by the BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of proteins (30 μg from each sample) were separated by 12 % SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5 % free-fat milk in TBS-T for 1 h at room temperature, and then incubated with primary antibodies against p65, p-p65 (1: 4,000) (Cell Signaling Technology, Beverly, MA, USA), iNOS (1: 1,000) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), or GAPDH with gentle agitation at 4 oC for overnight. After that, the membranes were incubated with secondary antibodies for 1.5 h at room temperature. After three washes with TBS-T, the membranes were developed using enhanced chemiluminescence (ECL, Thermo Scientific, Shanghai, China) and were then exposed to X-ray films. X-ray films were scanned and analyzed with ImageJ 1.41p software (National Institutes of Health, Bethesda, Maryland, USA) for protein quantification.
Statistical analysis
All results were presented as mean ± SD. Data were analyzed using a SPSS 13.0 statistical package. Multiple comparisons for data were performed by one-way ANOVA, followed by Dunnett’s test. A value of p < 0.05 was considered statistically significant.
Results
Effect of EE on H9c2 cell proliferation
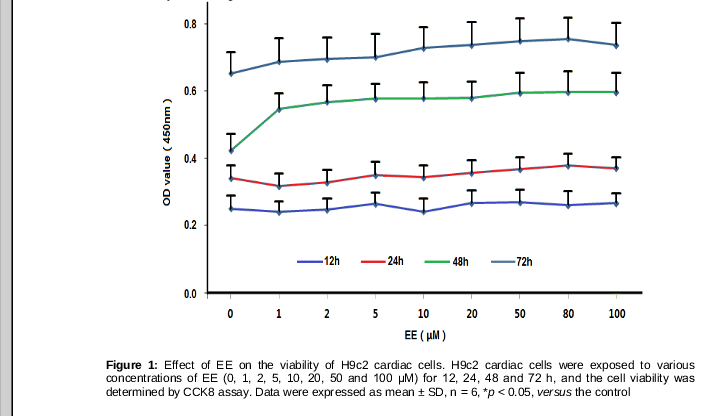
We first investigated the effect of EE on H9c2 cell proliferation. As shown in , EE promoted H9c2 cell proliferation in a time-dependent manner. Cell viability was notably elevated after being treated with 50-100 μM for 48h or 72h, while no significant difference was seen among these samples. Therefore 10, 50 and 80 μM of EE were used to carry out further investigations.
EE inhibits H9c2 cell apoptosis induced by DOX
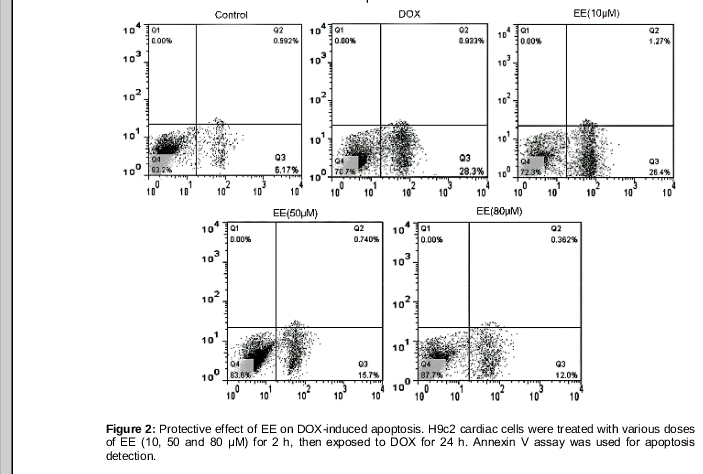
We then looked into whether or not EE influenced H9c2 cell apoptosis induced by DOX. The effect of EE on DOX-induced H9c2 cell apoptosis are shown in . DOX gave rise to higher rates of cell apoptosis in H9c2 cells (29.5 ± 3.56 %) compared with control group (6.39 ± 0.67 %. Pretreatment with EE (50 and 80 μM) for 2h decreased the apoptosis rates of the H9c2 cells to 16.8 ± 2.16 and 13.54 ± 2.08 %, respectively.
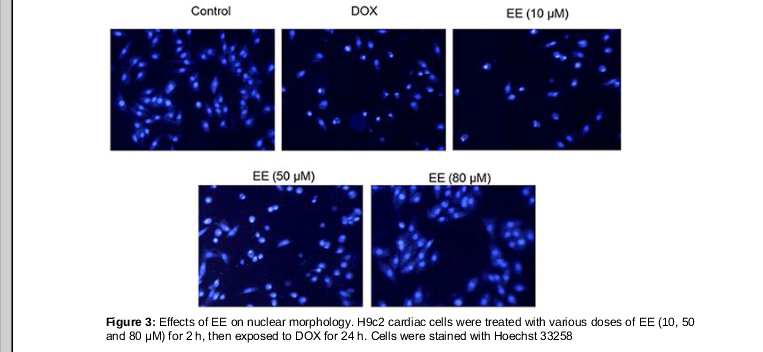
Morphological changes of H9c2 cells were observed with Hoechst 33258 staining (). Chromatin in normal cells showed a well-proportioned low density of fluorescence. H9c2 cells treated with DOX showed typical apoptosis characteristics with a concentrated compact density of fluorescence in the nucleus. The symptoms of apoptosis were significantly decreased in the EE pretreatment groups, indicating EE significantly inhibits H9c2 cell DOX-induced apoptosis of H9c2 cell.
EE suppresses IL-1β, IL-6 and TNF-α levels induced by DOX
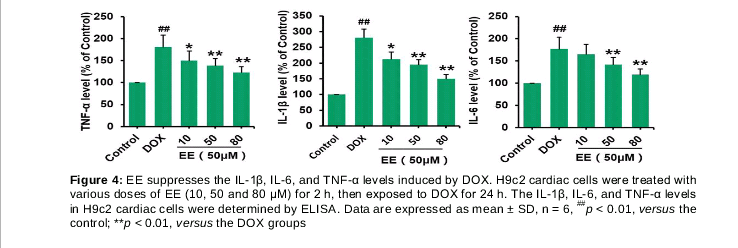
Next we examined EE effect on DOX-induced production of inflammatory cytokines. The levels of secreted IL-1β, IL-8, and TNF-α in the culture media were determined by ELISA. As shown in , when compared with control group, the levels of IL-1β, IL-6, and TNF-α in DOX treated groups were significantly increased by 174.5 ± 16.2, 74.9 ± 8.47 and 79.8 ± 9.61 %, respectively. However, 10, 50 and 80 μM of EE pretreatment remarkably reduced IL-1β and TNF-α levels in a dose-dependent manner, while 50 and 80 μM of EE pretreatment significantly decreased IL-6 level in compared with DOX treated groups.
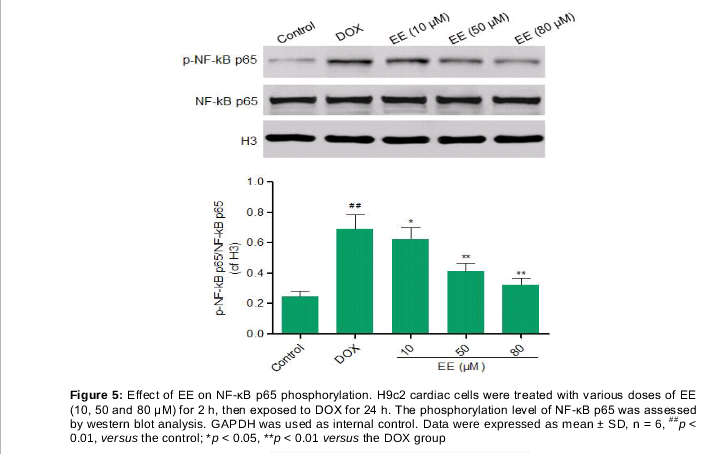
EE suppresses phosphorylation of NF-κB p65 induced by DOX
In order to elucidate the mechanism of protective effect of EE, we next examined the impact on phosphorylation level of NF-κB p65, which was detected by Western blot and the results are shown in . The phosphorylation level of NF-κB p65 in DOX group was increased by 181.22 ± 18.8 % compared with control group. EE pretreatment (10, 50 and 80 μM) remarkably reduced the phosphorylation of NF-κB p65 by 11.51 ± 1.25, 40.2 ± 5.17 and 52.97 ± 6.74 %, respectively.
EE attenuates DOX-induced apoptosis and inflammatory cytokine expressions in H9c2 cells via interaction with NF-κB signaling pathway
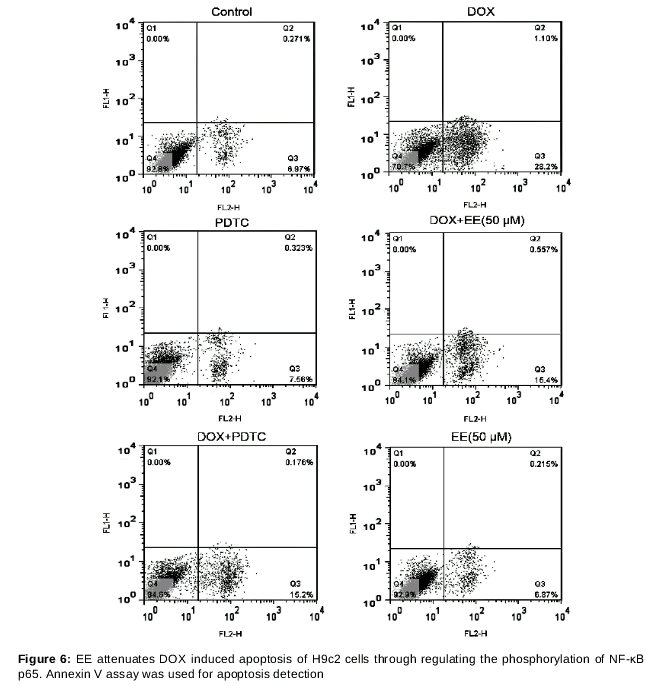
To further explore the mechanism by which EE alleviates DOX induced apoptosis and inflammation of H9c2 cells, H9c2 cells were pretreated with PDTC (a specific inhibitor of NF-κB) and EE (50 μM) prior to DOX treatment. As shown in , there was no significant difference in apoptosis rates between control group and PDTC group. Interestingly, while EE pretreatment (50 μM) effectively decreased H9c2 cell apoptosis in DOX+EE group as compared with DOX group, the apoptosis rate of the PDTC + DOX + EE group (25.2 ± 3.79 %) was comparable to that of DOX group (27.4 ± 3.56 %), suggesting that the effect of EE was much less effective in the presence of PDTC, therefore it can be concluded that the protective effect of EE is associated with NF-κB signaling pathway.
In addition, the levels of secreted IL-1β, IL-8, and TNF-α in the culture media was measured by ELISA. Similarly, no obvious difference in IL-1β, IL-8 and TNF-α levels was seen between PDTC + DOX + EE group and DOX group, indicating that EE failed to reduce the level of inflammatory cytokines induced by DOX in the presence of PDTC while it effectively decreased the level of inflammatory cytokines in DOX+EE group, as compared with DOX group.
Discussion
Our study demonstrated that EE promoted cell proliferation in a dose dependent manner and that EE pretreatment decreased the apoptosis rate and attenuated the inflammation of H9c2 cells induced by DOX treatment. Furthermore, the EE exerted its protective effect of on DOX-induced apoptosis and inflammation in H9c2 cells by regulating NF-κB signaling pathway.
DOX is reported to induce increases in the production of inflammatory cytokines such as IL-1β, IL-6 and TNF-α. In our experimental model, DOX also elicited an inflammatory response with an obvious increase in the production of IL-1β, IL-6, and TNF-α. EE could inhibit cytokine production produced by DOX, however this effect was greatly diminished inH9c2 cells pre-treated with PDTC, indicating that the phosphorylation of NF-κB p65 was responsible for the protecting role of EE on DOX-induced effects.
IL-1β is an initiator cytokine and plays a vital role in the regulation of inflammation response [15]. It has been shown to contribute to the accumulation of IL-6 and GCSF as well as cardiotoxicity induced by DOX.[16-18].
Recently, Zhu et al reported that the DOX treatment highly induced the expression levels of the IL-1β and IL-1 type 1 receptor (IL-1R1) and that the recombinant human IL-1 receptor antagonist (rhIL-1Ra) inhibited the DOX-induced acute cardiotoxicity in mice [19]. This suggests the involvement of IL-1β in DOX-induced cardiotoxicity. NF-κB activates COX-2 expression in response to multiple cytokines and growth factors, and participates in DOX-induced cardiomyocyte injury [20]. Thus, elucidating whether there is an interaction between IL-1β and the NF-κB pathway may lead to a method for ameliorating the inflammatory consequences of DOX.
In this study, we found that DOX-induced IL-1β expression and phosphorylation of NF-κB p65 expression decreased significantly with EE pretreatment, suggesting the involvement of the IL-1β in the activation of the NF-κB pathway. Combined with the result that pretreatment with PDTC inhibits the DOX-induced increase in IL-1β levels, it was revealed that there is a positive interaction between the NF-κB pathway and IL-1β during the development of inflammation and cytotoxicity induced by DOX. We further found that PDTC also decreases the apoptosis rate and the levels of secreted IL-1β, IL-6, and TNF-αin the culture medium. There is no significant difference between the PDTC and PDTC + EE (50 μM) treatments. The results suggest that the protective effects of EE on DOX induced apoptosis and inflammation contribute to the suppression of the phosphorylation of NF-κB p65. Therefore, EE can be employed as a new agent for the myocardial inflammation treatment.
Conclusion
The findings of this study suggested that EE decreased H9c2 apoptosis as well as the production of IL-1β, IL-6 and TNF-α induced by DOX by suppressing the phosphorylation of NF-κB p65. These findings may provide new insights into the development of novel therapies for myocardial inflammation.
References
Archives


News Updates